During my Ph.D., I pursued my interest in understanding how the structure and conformational dynamics of a biomolecule define its function in the computational biophysics laboratory of Dr. Nikolay Dokholyan. There, I applied modeling and molecular simulations to biological problems to generate insights that were then experimentally validated through collaborations with other research groups. I leveraged this approach in following major research projects:
Structural Distortion of DNA by Platinum Anticancer Drugs
Cisplatin and Oxaliplatin are two widely used platinum-containing anticancer drugs. Both these drugs bind and distort DNA, preventing proteins from reading the genetic code. However, cancer cells that are resistant to Cisplatin are still sensitive to Oxaliplatin. This is even more perplexing because the structure of Cisplatin bound to DNA looks exactly similar to the structure of Oxaliplatin bound to DNA. Because of the need to understand the differences between activity of Cisplatin and Oxaliplatin, we developed a simulation framework and performed molecular simulations to uncover the effect of the drugs on DNA flexibility and dynamics.
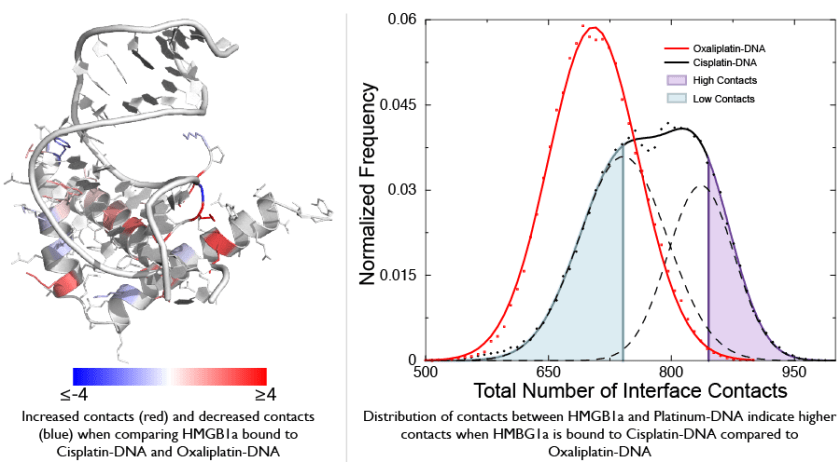
To find out how these two drugs could affect DNA differently other than the structure, we performed simulations that measured flexibility of DNA bound by both these drugs. These simulations show that DNA bound by Cisplatin is much more flexible compared to DNA bound by Oxaliplatin in one sequence context. This increased flexibility of Cisplatin-DNA compared to Oxaliplatin-DNA explained why Cisplatin-DNA was recognized by a protein called HMBG1 much better compared to Oxaliplatin-DNA. HMGB1 binds and distorts DNA, so if the DNA is more flexible it might bind better to HMGB1. Thus, our research uncovered a unique aspect of drugs that bind DNA – the effect of the drug on sequence-dependent DNA flexibility could profoundly alter the effect of the drug.
Publications
- Ramachandran, S., Temple, B., Alexandrova, A.N., Chaney, S. G., and Dokholyan, N. V., “Recognition of platinum-DNA adducts by HMGB1a”, Biochemistry 51(38):7608-17 (2012) pubmed journal
- King, C. L., Ramachandran, S., Chaney, S. G., Collins, L., Swenberg, J. A., deKrafft, K. E., Lin, W., Cicurel, L., and Barbier, M. “Debio 0507 primarily forms diaminocyclohexane-Pt-d(GpG) and -d(ApG) DNA adducts in HCT116 cells”, Cancer Chemotherapy and Pharmacology, 69(3):665-677 (2012) pubmed journal
- Bhattacharyya D.*, Ramachandran S.*, Sharma S., Pathmasiri W., King C. L., Baskerville-Abraham I., Boysen G., Swenberg J. A., Campbell S. L., Dokholyan N. V., and Chaney S. G. “Flanking bases influence the nature of DNA distortion by oxaliplatin 1,2-intrastrand (GG) cross-links” Public Library of Science One, 6:e23582 (2011) pubmed journal
- Ramachandran, S., Temple, B., Chaney, S. G., and Dokholyan, N. V. “Structural basis for the sequence dependent effects of Platinum-DNA adducts”, Nucleic Acids Research 37: 2434-2448 (2009) pubmed journal
Ryanodine Receptor Structure and Function
Ryanodine receptors (RyRs) are channels that provide a passage for ions through membranes of an intracellular calcium storage compartment called the sarcoplasmic reticulum. Nerve impulse triggers the opening of RyR channels, thus releasing calcium from the sarcoplasmic reticulum, which subsequently leads to muscle contraction. Congenital mutations in a specific type of RyR that is present in skeletal muscles, RyR1, lead to central core disease (CCD), which leads to weakened muscle. RyR1 mutations also render patients to be highly susceptible to malignant hyperthermia, an adverse reaction to general anesthesia. Although it is generally known that CCD mutations abort RyR1 function, the molecular basis of RyR1 dysfunction remains largely unknown because of the lack of atomic-level structure.
To address this crucial gap in our knowledge, we first built a structural model of the pore-forming region of RyR1 using cryoEM densities. Our structural model locates along the pore, the positions of residues critical for channel conductance and selectivity (the selectivity filter 4894 GGGIGDE 4900). Our structural model also shows preferential localization of Ca2+ over K+ near these important residues in molecular dynamics simulations, pointing to the basis for RyR1 Ca2+ selectivity observed in single channel measurements. Ca2+ selectivity due to selective binding at these residues was further validated by our simulations of RyR1-D4899Q and RyR1-G4898R mutants, which demonstrated a loss of preference to Ca2+ in the selectivity filter compared to loss of selectivity to Ca2+ observed experimentally.

Structural models of RyR1 in open and closed states. A, the experimental structures of Kv1.2 and MlotiK1 channels were used to construct the structural models of RyR1 in the open and closed states. The RyR1 models are displayed in the ribbon representation using PyMOL. B, the open state viewed from the luminal side and the cytoplasmic side featuring the overlay of cryo-EM densities (transparent surface) and the open state structural model (ribbon representation). The overlap of S6 and 45L with the corresponding traced densities is shown on the right. C, an overlay of the closed state structure and cryo-EM densities shown as viewed from the luminal side and the cytoplasmic side. The overlay of the S6 and 45L helices with the traced densities is shown on the right. The two views represent a 180° rotation around the vertical axis. The scale bars shown in the overlay of the structural models and EM densities measure 10 Å. D, the interface between 45L and S6 in the open and closed states (corresponding to the boxedregions A) is shown to highlight the higher conservation of the 45L-S6 interface compared with the rest of the regions. Each residue is colored according to its conservation derived from multiple sequence alignment of PSI-BLAST hits, with blue representing 100% conservation and redrepresenting 0% conservation. E, the residue propensities of 45L (left) and the C-terminal end of S6 (right) generated using the Berkeley WebLogo program.
We next generated structural models of the open and closed states of RyR1. Using our structural models, we identified an interface between the pore-lining helix (Tyr-4912–Glu-4948) and a linker helix (Val-4830–Val-4841) that lies parallel to the cytoplasmic membrane leaflet. To test the hypothesis that this interface controls RyR1 gating, we designed mutations in the linker helix to stabilize either the open (V4830W and T4840W) or closed (H4832W and G4834W) state. These mutants were generated and validated using single channel experiments at the Meissner lab. To further confirm this interface, we designed mutations in the pore-lining helix to stabilize the closed state (Q4947N, Q4947T, and Q4947S), which was also validated using single channel experiments at the Meissner lab. The channel conductance and selectivity of the mutations that I designed in the linker and pore-lining helices were indistinguishable from those of WT RyR1, demonstrating our ability to modulate RyR1 gating without affecting ion permeation. In summary, the combined theoretical and experimental studies have elucidated the ion conduction pathway and gating mechanism of RyR1.
Publications
- Shirvanyants, D., Ramachandran, S., Mei, Y., Xu, L., Meissner, G., Dokholyan, N. V. “Pore dynamics and conductance of RyR1 transmembrane domain”, Biophysical Journal, 106(11):2375-84 (2014) pubmed journal
- Ramachandran, S.*, Chakraborty, A.*, Xu, L.*, Mei, Y., Samso, M., Dokholyan, N. V., and Meissner, G. “Structural determinants of skeletal muscle ryanodine receptor gating”, Journal of Biological Chemistry, 288(9):6154-65 (2013) pubmed journal
- Ramachandran, S., Serohijos, A. W. R., Xu, L., Meissner, G., and Dokholyan, N. V. “A structural model of the pore-forming region of the skeletal muscle Ryanodine receptor (RyR1)”, Public Library of Science Computational Biology 5: e1000367 (2009) pubmed journal
- Meissner, G., Pasek, D. A., Yamaguchi, N., Ramachandran, S., Dokholyan, N. V., and Tripathy, A. “Thermodynamics of calmodulin binding to cardiac and skeletal muscle ryanodine receptor ion channels”, Proteins: Structure, Function, and Bioinformatics, 74: 207-211 (2009) pubmed journal
